It is easy to draw molecules like benzene with an external structure
editor but molecules suitable for use in functional groups is more difficult.
An external molecule editor is required!
Any free or commercial structure editor can be used to create molecule
files in the *.mol file format.
Functional groups are meant to be parts of such molecules. From the
functional group point of view, it is a molecule residual. In the interpretation
rule database these molecule fragments are used to identify a functional
group within an analyzed molecule. For this purpose,
the residual part a molecule fragment is attached to needs to be well
defined according to the allowed chemical environment.
Example:
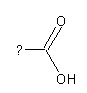
A carboxylic acid functional group is shown in the following, which
might be attached to any residual molecule:
In the corresponding functional group definition, the molecular
fragment needs to be assigned. Furthermore, the unknown residual (indicated
by the question mark) needs to be specified in more detail. This is useful
to distinguish e.g. aromatic from aliphatic carboxylic acids. In case
of aromatic carboxylic acids, the residue must be an aromatic compound,
but it would never be any aliphatic compound like an Alkyl-group. Therefore,
residuals (indicated by the question mark) will be considered as Generic
atoms with special properties. These properties can be defined in the
atom property dialog (see below).
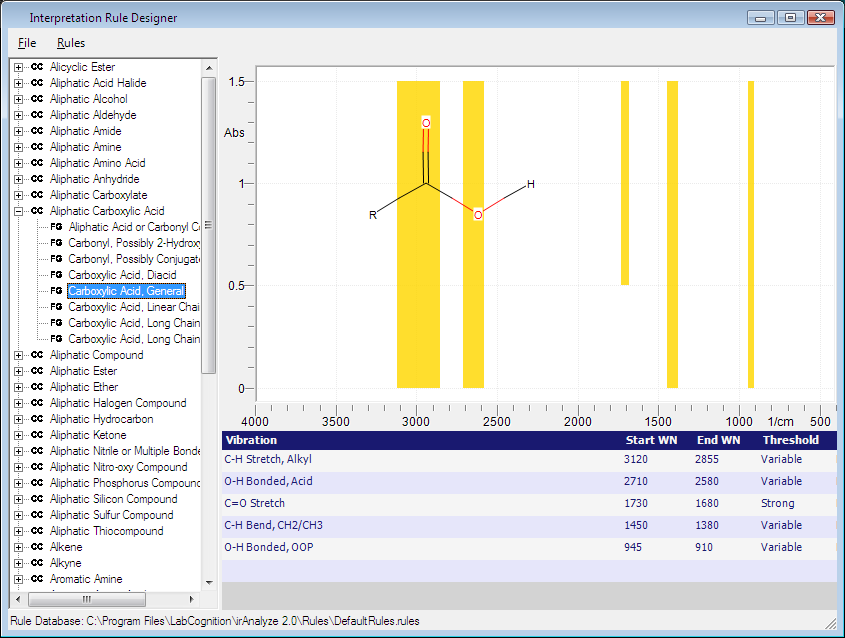
Workflow for Designing a Functional Group Molecule
To design a functional group molecule, please follow the instructions
below:
Open the external
structure editor and draw the functional group of interest.
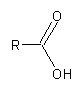
Replace any open residual by an R atom.
Example:
Carboxylic acid molecular fragment
Save or export the drawn molecule file (*.mol
or *.sk2).
Select the destination
functional group node in the tree of the Rule Designer.
From the MS-Windows explorer, drag the molecule file
(*.mol or *.sk2) and drop it onto the molecule section of the Rule
Designer:
Double click
with the Left
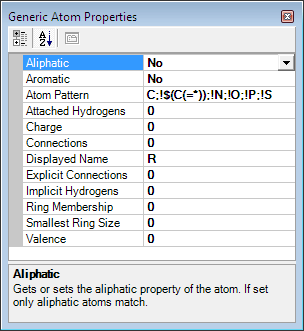
mousebutton onto the R atom within the molecule view to edit
the Generic atom properties.
An Atom Properties dialog is opened, where generic atom
properties according to the SMILES and SMARTS nomenclature can be adjusted.
Please refer to the "Generic
Atom Properties" section for details.
After modifying the atom properties, close
the dialog clicking the button.